
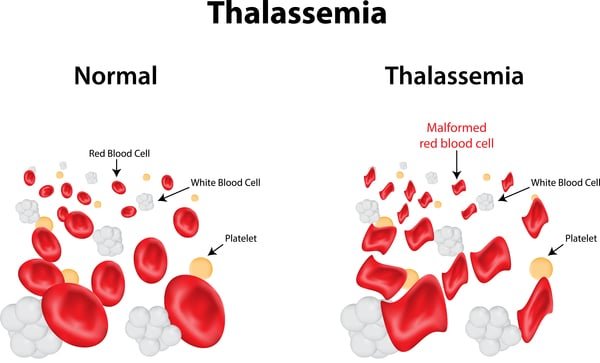
thalassemia Disease is a genetic blood disorder characterized by less production of hemoglobin and fewer red blood cells in the body than normal. Hemoglobin is the protein in red blood cells that carries oxygen from the lungs to the rest of the body. The condition can cause various health problems, ranging from mild to severe anemia. There are two main types of thalassemia: alpha thalassemia and beta thalassemia, depending on which part of the hemoglobin molecule is affected.
Types of thalassemia Disease
Alpha Thalassemia:
Cause: Results from mutations or deletions in the alpha-globin genes on chromosome 16.
Severity: Can range from silent carrier state, minor anemia, hemoglobin H disease, to the more severe alpha thalassemia major (hydrops fetalis).
Beta thalassemia Disease:
Cause: Results from mutations in the beta-globin genes on chromosome 11.
Severity: Includes beta thalassemia minor (carrier state), beta thalassemia intermedia, and the severe beta thalassemia major (Cooley’s anemia).
Causes
Thalassemia is inherited in an autosomal recessive pattern, meaning both parents must be carriers of the mutated gene to pass it on to their children. If both parents are carriers, there is a 25% chance with each pregnancy that their child will have thalassemia major, a 50% chance the child will be a carrier, and a 25% chance the child will have normal hemoglobin.
Symptoms
The symptoms of thalassemia vary depending on the type and severity of the disease:
Mild Thalassemia (carrier state): May have mild anemia, often without noticeable symptoms.
Moderate to Severe Thalassemia: Symptoms include severe anemia, fatigue, weakness, pale or yellowish skin (jaundice), facial bone deformities, slow growth, abdominal swelling, and dark urine.
Diagnosis
Thalassemia can be diagnosed through various tests:
Complete Blood Count (CBC): To check for low red blood cell count and abnormal hemoglobin levels.
Hemoglobin Electrophoresis: To identify abnormal types of hemoglobin.
Genetic Testing: To detect mutations in the alpha or beta-globin genes.
Prenatal Testing: Amniocentesis or chorionic villus sampling (CVS) to diagnose thalassemia in the fetus if both parents are carriers.
Treatment
Treatment for thalassemia depends on the severity of the condition:
Regular Blood Transfusions: Necessary for patients with severe thalassemia to maintain adequate hemoglobin levels.
Iron Chelation Therapy: Removes excess iron from the body caused by frequent blood transfusions. Common chelating agents include deferoxamine (Desferal), deferiprone (Ferriprox), and deferasirox (Exjade, Jadenu).
Folic Acid Supplements: Help in the production of red blood cells.
Bone Marrow or Stem Cell Transplant: Can potentially cure thalassemia, especially in children with a suitable donor. This procedure involves replacing the defective bone marrow with healthy marrow from a compatible donor.
Gene Therapy: Emerging treatments involving the correction of genetic defects causing thalassemia are under research and development.
Living with thalassemia Disease
Patients with thalassemia should take specific measures to manage their condition and improve their quality of life:
Regular Medical Follow-ups: Monitoring hemoglobin levels, iron levels, and organ function.
Healthy Diet: A balanced diet rich in vitamins and low in iron to avoid excess iron accumulation.
Avoiding Infections: Regular vaccinations and prompt treatment of infections.
Psychological Support: Counseling and support groups to cope with the emotional and psychological challenges of living with a chronic illness.
Preventive Measures
Genetic counseling is crucial for at-risk couples (where both partners are carriers of the thalassemia gene) to understand the risks and consider options such as prenatal diagnosis or in vitro fertilization (IVF) with preimplantation genetic diagnosis (PGD) to prevent the birth of affected children.
Thalassemia is a complex genetic disorder requiring lifelong management. Advances in medical treatment, early diagnosis, and supportive care have significantly improved the outlook for patients with thalassemia. With appropriate care, individuals with thalassemia can lead fulfilling lives. Public awareness, genetic counseling, and ongoing research into innovative therapies remain essential to further enhance the quality of life for those affected by this condition.
Causes of thalassemia Disease
Thalassemia is a hereditary blood disorder caused by mutations in the genes responsible for the production of hemoglobin, the protein in red blood cells that carries oxygen throughout the body. The primary cause of thalassemia is genetic mutations, which are inherited in an autosomal recessive manner. Here’s a detailed look at the causes:
Genetic Mutations
Thalassemia occurs due to mutations or deletions in the globin genes responsible for producing hemoglobin. There are two main types of thalassemia, each associated with different genetic mutations:
Alpha thalassemia Disease:
Cause: Resulting from mutations or deletions in the HBA1 and HBA2 genes on chromosome 16, which are responsible for producing the alpha-globin chains of hemoglobin.
Inheritance: Each person has four alpha-globin genes (two from each parent). The severity of alpha thalassemia depends on the number of affected genes:
Silent Carrier: One gene is mutated; usually no symptoms.
Alpha Thalassemia Trait: Two genes are mutated; mild anemia.
Hemoglobin H Disease: Three genes are mutated; moderate to severe anemia.
Alpha Thalassemia Major (Hydrops Fetalis): All four genes are mutated; usually fatal before or shortly after birth.
Beta thalassemia Disease:
Cause: Resulting from mutations in the HBB gene on chromosome 11, which is responsible for producing the beta-globin chains of hemoglobin.
Inheritance: Each person has two beta-globin genes (one from each parent). The severity of beta thalassemia depends on the number of mutated genes:
Beta Thalassemia Minor (Carrier): One gene is mutated; mild anemia.
And Beta Thalassemia Intermedia: Two genes are mutated but with less severe mutations; moderate anemia.
Beta Thalassemia Major (Cooley’s Anemia): Two severely mutated genes; severe anemia requiring regular blood transfusions.
Inheritance Pattern
Thalassemia is inherited in an autosomal recessive pattern. This means that both parents must carry and pass on the mutated gene for their child to have thalassemia major. If both parents are carriers (thalassemia minor), the inheritance pattern is as follows for each pregnancy:
25% chance: The child will have thalassemia major (inherits two mutated genes).
50% chance: The child will be a carrier (inherits one mutated gene).
25% chance: The child will have normal hemoglobin (inherits no mutated genes).
Geographic and Ethnic Factors
Thalassemia is more prevalent in certain parts of the world due to historical resistance to malaria conferred by the carrier state:
Mediterranean Region: Including countries like Greece, Italy, and Cyprus.
Middle East: Including countries like Saudi Arabia, Iran, and Jordan.
South Asia: Including countries like India, Bangladesh, and Pakistan.
Africa: Particularly in sub-Saharan regions.
The high frequency of thalassemia carriers in these regions is due to natural selection, where carriers have a survival advantage against malaria.
Thalassemia is caused by genetic mutations affecting the production of hemoglobin. The severity of the disease depends on the type and number of mutations inherited. Understanding the genetic basis and inheritance patterns of thalassemia is crucial for diagnosis, management, and prevention. Genetic counseling and prenatal testing are important tools for at-risk couples to make informed decisions about family planning.
Treatment Methods for thalassemia Disease
Thalassemia is a genetic blood disorder that requires comprehensive and ongoing treatment to manage symptoms and prevent complications. The choice of treatment depends on the type and severity of the thalassemia. Here are the primary treatment methods:
1. Regular Blood Transfusions
Purpose: To maintain adequate hemoglobin levels and reduce anemia.
Frequency: Patients with severe thalassemia (such as beta thalassemia major) may require transfusions every 2-4 weeks.
Considerations: Regular transfusions help to ensure proper oxygen delivery to tissues and support normal growth and development in children.
2. Iron Chelation Therapy
Purpose: To remove excess iron from the body that accumulates due to frequent blood transfusions.
Medications:
Deferoxamine (Desferal): Administered via subcutaneous infusion using a small pump over 8-12 hours, several times a week.
Deferiprone (Ferriprox): Taken orally, typically multiple times a day.
Deferasirox (Exjade, Jadenu): Taken orally, once daily.
Side Effects:
Deferoxamine: Injection site reactions, hearing loss, vision problems.
Deferiprone: Nausea, vomiting, joint pain, neutropenia (low white blood cell count).
Deferasirox: Gastrointestinal issues, increased liver enzymes, kidney problems.
3. Bone Marrow or Stem Cell Transplant
Purpose: To replace the defective bone marrow with healthy stem cells capable of producing normal hemoglobin.
Procedure:
Donor Selection: Requires a compatible donor, often a sibling.
Pre-transplant Preparation: Includes chemotherapy or radiation to destroy the patient’s existing bone marrow.
Transplant: Infusion of healthy stem cells from the donor.
Considerations: This procedure carries significant risks, including graft-versus-host disease (GVHD), infections, and organ damage, but it can be curative for some patients.
4. Medications to Increase Fetal Hemoglobin Production
Hydroxyurea:
Purpose: To stimulate the production of fetal hemoglobin (HbF), which can reduce the severity of anemia.
Administration: Taken orally.
Side Effects: Bone marrow suppression, gastrointestinal disturbances, increased risk of infections.
5. Folic Acid Supplements
Purpose: To support the production of red blood cells and reduce anemia.
Administration: Taken orally, typically as a daily supplement.
Side Effects: Generally well-tolerated, but high doses can cause gastrointestinal discomfort.
6. Gene Therapy (Experimental)
Purpose: To correct the genetic defect causing thalassemia.
Approaches:
Gene Addition: Introducing a functional copy of the defective gene.
Gene Editing: Directly correcting the mutation within the patient’s DNA.
Status: Currently under clinical trials and research, with promising but still experimental results.
7. Supportive Care
Regular Monitoring: Frequent blood tests to monitor hemoglobin levels, iron levels, and organ function.
Vaccinations: To prevent infections, especially important if the spleen is removed (splenectomy).
Psychosocial Support: Counseling and support groups to help patients and families cope with the emotional and psychological impacts of the disease.
Conclusion
Managing thalassemia requires a multidisciplinary approach involving regular medical care, adherence to treatment protocols, and supportive therapies to improve quality of life. Advances in medical research continue to explore new treatments, including gene therapy, which may offer hope for a cure in the future. Collaboration between patients, healthcare providers, and caregivers is essential for optimal management of this complex condition.
Commons questions about thalassemia Disease
here are some common questions about thalassemia along with their answers :
What is thalassemia?
Answer: Thalassemia is an inherited blood disorder that affects the production of hemoglobin in the blood, leading to chronic anemia.
What are the types of thalassemia?
Answer: There are several types of thalassemia, including thalassemia major, thalassemia minor, and other variants such as thalassemia intermedia.
How is thalassemia diagnosed?
Answer: Thalassemia is typically diagnosed through blood tests to measure hemoglobin levels and examine red blood cells.
What are the common symptoms of thalassemia?
Answer: Symptoms vary depending on the type and severity of thalassemia, but they may include anemia, fatigue, headache, shortness of breath, and chest pain.
Can thalassemia be treated?
Answer: There is no cure for thalassemia, but symptoms can be managed, pain can be alleviated, and the risk of complications can be reduced through medication, blood transfusions, and other treatments.
Can thalassemia be prevented from being passed on to offspring?
Answer: The risk of passing on thalassemia can be reduced through genetic testing before marriage and consulting with genetic counselors.
These are some common questions about thalassemia along with their answers. Individuals suspected of having thalassemia should consult a specialist for more accurate information and appropriate treatment guidance.
websites that provide comprehensive information about thalassemia Disease
Here are some reliable websites that provide comprehensive information about thalassemia Disease :
Centers for Disease Control and Prevention (CDC) – Offers detailed information on thalassemia, including symptoms, complications, and risk factors. Visit CDC.
National Heart, Lung, and Blood Institute (NHLBI), NIH – Covers various aspects of thalassemia such as what it is, its symptoms, diagnosis, treatment, and living with the condition. Visit NHLBI.
Cooley’s Anemia Foundation – Focuses on beta thalassemia and provides information on types, symptoms, and treatment options. Visit Cooley’s Anemia Foundation.
MedlinePlus – A service of the National Library of Medicine (NLM) that offers information about thalassemia including its genetics, diagnosis, and management. Visit MedlinePlus.
Britannica – Provides an overview of thalassemia, its pathology, diagnosis, and prevention. Visit Britannica.
These sources are trustworthy and provide a wide range of information on thalassemia for both patients and healthcare providers.